Executive Summary
Across the week, presentations and discussions revealed the latest strategies and processes driving transformation of pharmaceutical regulatory affairs around the world.
From e-Submissions and harmonisation across different regions to RIMS and optimising data management, the sessions covered a wide range of hot topics.
Medical and Regulatory Researcher and Writer Zoya Marinova provides an overview of the three main themes addressed by top presentations in each conference track.
Regulatory reform and the drug approval process in China: where are we now?
Ching Li, Regulatory Manager, Biotest Pharma GmbH
Ching Li described the modernisation of the regulatory approval process in China since the amendment of the Drug Administration Law and the implementation of the new vaccine administration law on 1st December 2019. Recent changes include provisions for drug registration regulation (DRR) and for the supervision and administration of drug production GMP, a draft for variation guidelines, and a pilot programme for breakthrough, conditional, and priority regulatory procedures. Further expected milestones include the drug category submission requirements for CTD and the Chinese Pharmacopeia.
The Drug Administration Law was first enacted in China in 1984 and underwent its second major amendment in 2019. It has been expanded into 155 articles in 12 chapters and aims to establish a scientifically sound and strict supervision and administration system, to promote risk management, and to ensure drug safety and quality. The amended law focuses on innovation, encourages clinical value-oriented R&D, and regulates the overall process of management and control (GxP).
Li then explained how the revised drug registration regulations address the implementation of drug innovation. Applicants have been expanded to R&D entities that can bear the corresponding legal responsibility. Moreover, the responsibilities of national and provincial regulatory agencies have been clarified, the regulation has been brought in line with international rules, and the review and approval process has been optimised. The adherence of the Center of Drug Evaluation (CDE) to these timelines was illustrated. In 2019, 90% of the applications were reviewed and approved by the CDE on time, and the backlog was reduced greatly. The number of applications to the CDE until July 2020 has increased by 15.4% compared to the same period in 2019, showing that the pandemic did not slow down the application process. Moreover, the CDE demonstrated the capacity to work efficiently under these exceptional circumstances.
Finally, Li showed how the pilot regulatory pathways for breakthrough designation, priority review, conditional approval, and exceptional circumstances function to encourage innovation. The priority pathway has received most applications with a focus on products with obvious clinical value that meet an urgent medical need and show clear advantages.
Regulatory strategy in EAEU and rest of CIS countries: points to consider for successful market access
Nargiz Asgarova, BSc, MBA, Regulatory Affairs Projects Manager, Biomapas
Nargiz Asgarova presented regulatory strategies for successful market access in the Commonwealth of Independent States (CIS) countries, focusing on the current situation, challenges, practical tips, and GMP requirements. Her presentation covered both the Eurasian Economic Union (EAEU) and NON-EAEU CIS countries.
The EAEU, which includes Armenia, Belarus, Kazakhstan, Kyrgyzstan, and Russia, is moving toward a process of common EAEU Marketing Authorization Applications (MAAs). Beginning 1st January 2021, all new MAAs should be submitted according to the common EAEU information electronic portal (CIEP). However, national Marketing Authorizations (MAs) will remain valid until 2025.
Challenges currently faced by the EAEU include the interaction between HAs, lack of clear guidance for confidential information, submission of variations with RVP, different national duration of data exclusivity, and harmonisation of switching to the EAEU normative document requirements and upcoming EAEU Pharmacopeia. As of August 2020, there are 78 EAEU MAs. The COVID pandemic also poses a challenge by impacting GMP inspection procedures and clinical studies conductance.
The next focus of the presentation was the NON-EAEU CIS countries, including Azerbaijan, Georgia, Moldova, Mongolia, Tajikistan, Turkmenistan, Ukraine, and Uzbekistan. The differences in the regulatory processes of the countries were highlighted. In 2020, different regulatory updates have been carried out in Azerbaijan, Ukraine, and Uzbekistan.
The national MAA timelines vary among the NON-EAEU CIS countries between 3 months and 15 months for the standard procedure and between 1 month and 6 months for the accelerated procedure. The accelerated procedure offers many advantages that may include a simplified dossier, a dossier identical to the reference country, reduced HA fees, or shorter review timelines. Challenges encountered during the regulatory process in the NON-EAEU countries include differences in the specific national regulations, the changing regulatory environment, different labelling requirements, or high-level requirements.
Finally, Asgarova described the GMP requirements in CIS countries. The EAEU is transitioning from national GMP certificates toward EAEU GMP certificates. The EAEZ CGM certification procedure includes a GMP inspection at the manufacturing site for compliance with EAEU GMP requirements, which should be obtained prior to the MAA or RVP and is valid for 3 years.
The fusion of IDMP into submission data
Dr Anna Thaidigsmann, Senior Business Consultant, EXTEDO
Dr Anna Thaidigsmann began her presentation by showing how the implementation of xEVMPD in 2014 has contributed to the introduction of DATA Only submissions and has served as a basis for PV fee calculations. She illustrated the business processing model for document submission and the role of xEVMPD in this process.
Her presentation showed that we are at a timepoint of increased fusion of IDMP and submission data, and the need for data exchange is boosted by a number of EU initiatives. Particularly, the IDMP serves as a link between the unstructured data of the SmPC and the semi-structured data of the eAF.
Defined, well controlled vocabularies (CVs) serve as the basis for DATA Only submissions. The role of CV usage in MAH with IDMP data sets, eAF, and backbone and content plan creation was illustrated, and an example of a real-life CV IDMP fusion was presented. Notably, different databases with high value sets may exist within a company. In these cases, business continuity and high data quality should be ensured before the fusion of data sets.
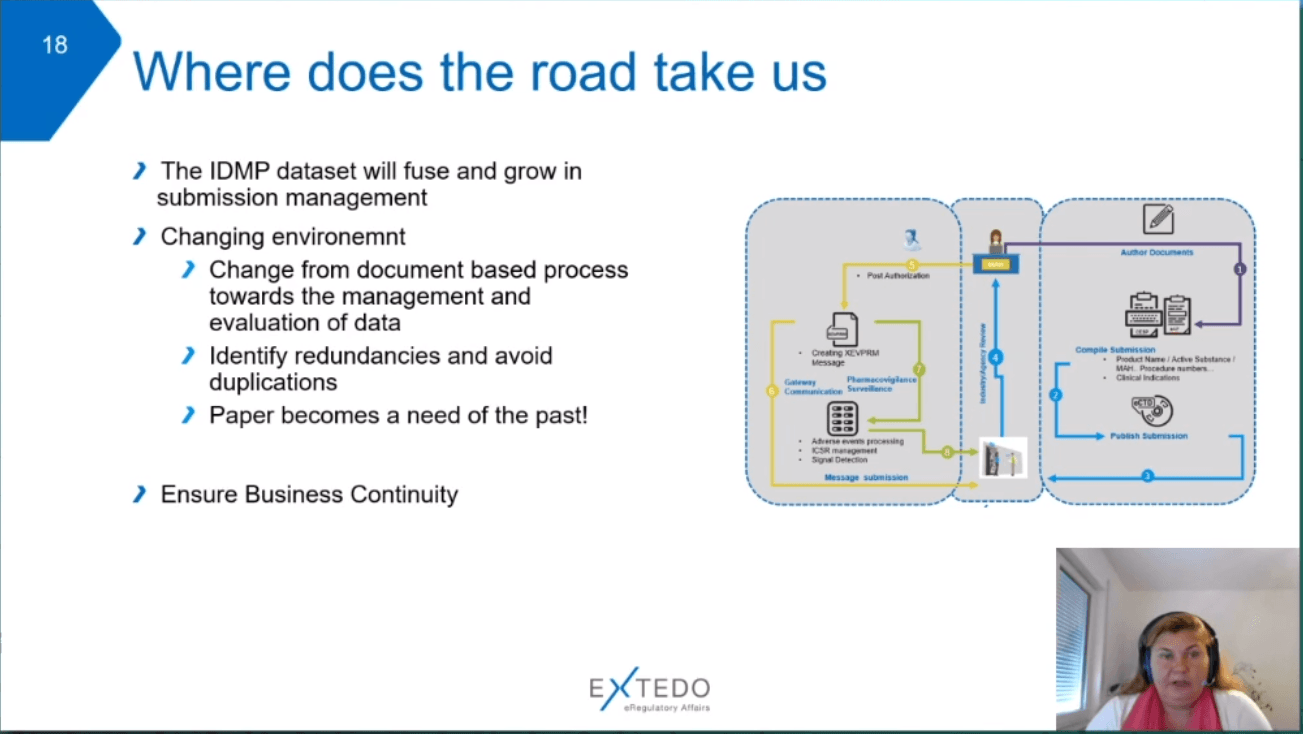
Finally, Dr Thaidigsmann summarised the key points of her presentation. The IDMP dataset will fuse and grow in submission management. In the changing environment, there is a focus on shifting from document-based processes toward the management and evaluation of data while ensuring data continuity.
Vision of an integrated submission landscape
Laurent Lefebvre, Associate Director in Regulatory Affairs Global Drug Development CMC, Novartis
Laurent Lefebvre presented his vision of an integrated submission landscape. He first reviewed how the industry has progressively added data aspects to the document-exchange based regulatory approach in order to create a holistic regulatory information management system. He envisions the creation of this type of integrated system also on the HA side to facilitate submission, dossier, and data exchange.
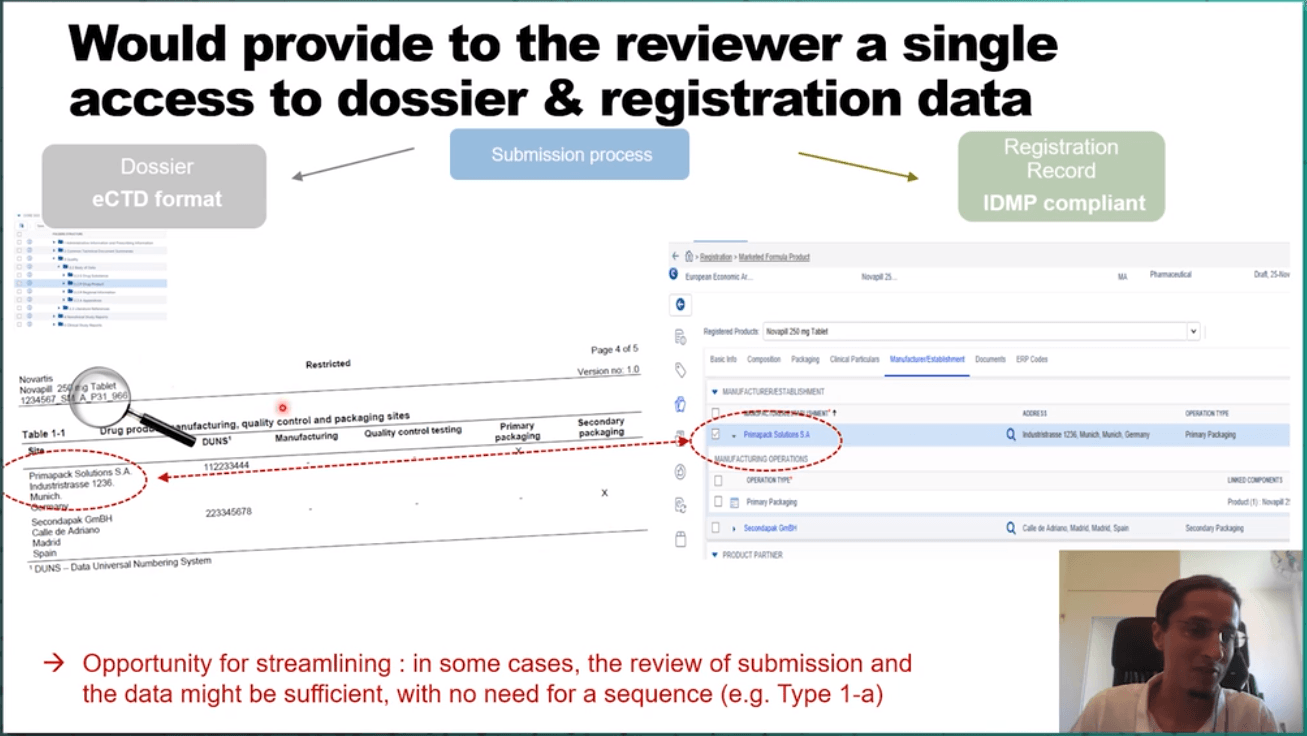
Challenges encountered in the communication with HAs include the separate submission, dossier, and registration management. In contrast, the industry trend is to assemble all regulatory submission elements in a single place integrating all capabilities – a RIM system compliant with the IDMP format. Lefebvre then presented an existing industry RIM to illustrate the interconnectedness of the RIM elements. The vision aligned to the regulatory process will include an interactive exchange of questions and responses and the modification by each data owner of their information.
The presented long-term vision for the IDMP landscape will fundamentally change the way information is exchanged and will streamline the business process. Lefebvre proposes to create a cloud-based EU RIM platform. It can connect the industry RIMS and a cloud-based EU HA RIM to share submission, dossier, and registration information. This would provide the reviewer a single access to dossier & registration data and streamline the process. Overall, this would create a dynamic review/approval process through direct messaging between RIMs.
Enhancing the overall efficiency of global Regulatory Information Management
Raj Srinivasan, Executive Business Partner, and Chikkam Ram Mohan Rao, Global Regulatory Business Partner, Navitas Life Sciences
This presentation focused on three main topics: challenges the industry faces with RIM management, optimisation of data handling across multiple regulatory systems, and process and data integration of end-to-end RIM.
Over the last decades, RIM has evolved from a paper process to digitalisation, exchange of documents, and finally exchange of data.
The driving forces behind this evolution are the harmonisation of data standards, collaboration among regulatory bodies, and the traceability and transparency of data.
A critical factor in this process is the change from application-centric to data-centric mindset to enable a single source of truth, alignment, and connectivity across all functional areas for transparency and accuracy. The data-centric approach considers data as a primary and permanent asset and builds a culture and architecture that puts data at the core of the organisation.
The strategic, business value, and operational RIM priorities explore data centricity, progress the RIM culture, and implement RIM, respectively. Data centricity is driven by the business, and an approximately 55% of all companies have started their journey to becoming data-centric. The “5x5x5” approach for progressing the RIM culture was presented, which considers 5 dimensions (governance, process, culture, data, and technology) and 5 RIM maturity levels.
Regulatory affairs should support science and discovery, regulatory information data provisioning, structured submissions, connectivity and interoperability, and life cycle management. A case study was presented for the establishment of an efficient RIM that improved data management and data quality, mitigated delays, ensured global alignment and transparency for timely change implementation, and resulted in zero post-approval compliance issues.
The RIM process flow includes internal and external stakeholders and has become more complex with the evolving regulatory requirements. Data handling across multiple regulatory systems should be facilitated to ensure an integration and optimisation of the process.
ISO IDMP related implementation at and by the regulator - MEDB experience and views on substance management
Joris Kampmeijer, CIO & Frits Stulp Project Manager EU-SRS, CBG-MEB Netherlands
Substance management is a critical subject for regulators. An example is the identification of the possible human carcinogens nitrosamines in sartan medicines in 2018.
This led to a full review of the procedures for substance management, and further development of information technology systems (including for substances) was recommended. In that regard, the European Substance Registration System (EU-SRS) is crucial to support substances (S), veterinary and human medicinal products (P), organisations (O), and referentials (R) (SPOR).
The scientifically sound EU-SRS system was originally developed by the FDA. It includes hierarchy and relationships between substances, references, codes and names, chemical structures, and search functions. The EU-SRS has been endorsed for implementation during 2020–2022. Moreover, it has been included in the Telematics Strategy & Roadmap and in the UNICOM project, which will focus on the conversion of key regulatory and clinical processes to use IDMP. By accelerating the diffusion of ISO IDMP standards, UNICOM supports regulatory processes, cross-border digital health services, global pharmacovigilance, and improved healthcare and medical research. Currently, a lot of effort is focused on the implementation and validation of the EU-SRS, which will be handed over to the EMA in 2022.
Next, the presenters reviewed the role of EU-SRS in SPOR and in reaching the ultimate goal: “to have one version of the truth”. To attain this goal, structured data and supporting documents should be submitted, the structured data should be assessed, and the approved data should be stored in an EU-database. However, this is not the current standard procedure, and a process is being developed to move from post-authorisation to pre-submission of structured data. The SPOR Task Force endorses a two-phase approach, building upon a centralised implementation first (EMA) and allowing the transition of NCAs to ISO IDMP over a period of time.
Current goals of the overall programme management of this part of the Telematics Roadmap include pre-registration of required master data to support various processes, product assessment and approval, signal management using coded data, clinical trial management, reporting of shortages, etc. This process is based on a common data repository of product information in Europe. Industry can also contribute by preparing IDMP/SPOR data on products, upgrading processes and systems where needed, contributing to the massive change in the joint way of working with HAs, and staying open and constructive.
Telematics Strategy in the EU
Vada A Perkins, Executive Director, Regulatory Policy & Intelligence/Head, Regulatory Intelligence, Bayer
Vada Perkins reviewed the impact of telematics globally and specifically in the EU and highlighted certain perspectives regarding the international market. He began his presentation with the traditional definition of telematics, which is the long-distance transmission of computerised information. However, he stressed that the role of telematics has evolved with the data revolution in regulatory science, which has placed high value on data integrity.
As a result, data governance, guiding principles, and data standards are being developed by international regulators and industry to ensure a transition to more efficient regulatory assessment. An example of quality standards implementation is the FDA cross-centre initiative for structured PQ/CMC submissions by the development of standardised data elements, terminologies and data structures, and the implementation of a data exchange standard for PQ/CMC data. This initiative has recently been internationally endorsed as a new topic for the ICH.
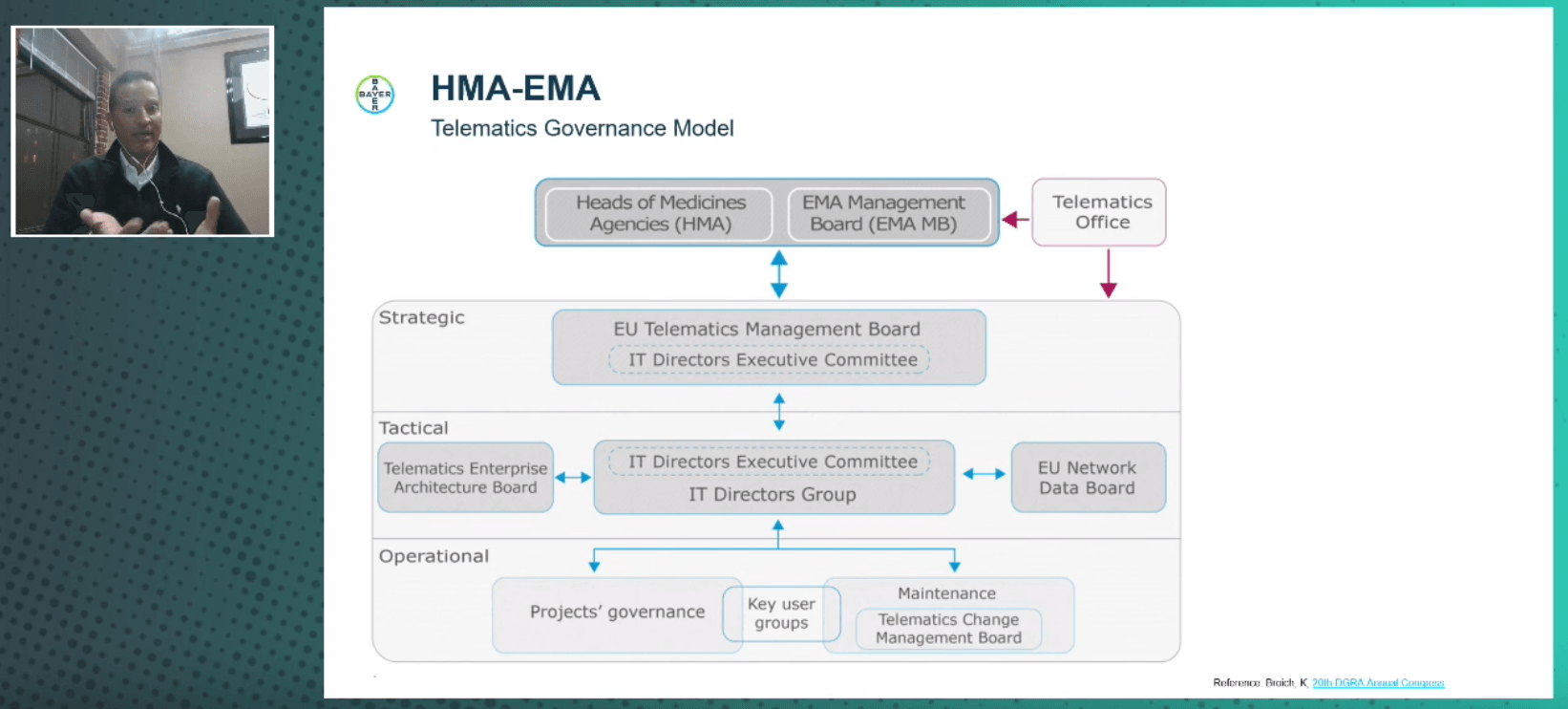
Next, Perkins presented the telematic governance model of the HMA-EMA. Its strategic efforts are being led by the EU Telematics Management Board. It includes several relevant programmes, such as eCollaboration, Pharmacovigilance, Clinical Trials, Data Integration, and Veterinary. The EU IDMP/SPOR Task Force Group falls under the Data Integration programme. However, the Extended Telematics Implementation Roadmap 2020–2021 also impacts a number of other programmes and standards.
The EU HMA/EMA Joint Big Data Taskforce has provided 47 recommendations and prioritisation of future actions. The aim is to achieve evidence acceptability in support of the evaluation and supervision of medicines in nine key areas. Notably, two of these nine areas are data standardisation and data quality, which emphasises the pivotal role of data quality for data analytics.
The EU Telematics Strategy Concept Paper 2020–2025 sets the aims to streamline regulatory decision making through master data management, sharing/exchange of assessment reports, and technical standards to facilitate automated data and document exchange and to reduce the administrative burden on data management.
Perkins stressed that the concept of telematics is evolving, and telematics is now being viewed as a horizontal activity to support a broad range of initiatives. He then illustrated how telematics is relevant to different focus areas of the European medicines regulatory network (EMRN) Strategy 2025. Telematics is pertinent to the availability and accessibility of medicines (Focus Area 1) by matching supply data and demand data of medicinal products at a network level; reducing the barriers to national access or distribution via electronic public information, and increasing the transparency on the marketing status of centrally authorised medicines and the insight to what is marketed in neighbouring countries.
Telematics is also relevant to the strategic goals of Focus area 2, including digital health, real world data, data and process analytics, digital tools, and digital transformation. Telematics can contribute to innovation (Focus area 3) by providing digital tools and data standardisation and supporting innovation and digitalisation in clinical trials.
Notably, telematics is especially relevant to Focus area 6 (sustainability of the Network and operational excellence) to drive regulatory optimisation, ensure that all NCAs contribute to Network operations, and combine and enrich EMRN internal data with external data sources.
Perkins also addressed the challenges associated with the telematics relevance to the EMRN Strategy 2025, including resource allocation and funding gaps, and strategies to overcome them. He concluded his presentation by emphasising the need to think holistically in redefining the role of telematics, taking into account the development of technology, IT, and regulatory sciences.
Following his presentation, Perkins answered several questions. Regarding the chances of Europe to be a leader in the digitalisation of the regulatory process, he emphasised that both cloud and dynamic regulatory assessment initiatives are currently being discussed in Europe and other areas and will be important for the advancement of the field. The challenges faced by small and medium enterprises in terms of digitalisation affordability were also addressed.
Perkins believes that accommodations are necessary for small and medium enterprises to ensure they can have access to digitalisation. Moreover, the involvement of multiple stakeholders from health authorities, industry, and data and technology companies in the process is important because it will make digitalisation more ubiquitous and bring down the cost.
The role of the industry in the Telematics Strategy implementation was discussed. Perkins believes that all stakeholders involved in the life cycle of data should be involved in the process, and the role of industry as a data generator is very important. Moreover, there will be a need for even broader cooperation between regulatory life sciences and healthcare. The role of the WHO as an important stakeholder in the process was also addressed.